For US Healthcare Professionals Only
Safety

For adults with relapsed or refractory MLNs with FGFR1 rearrangement
A targeted therapy with a well-established safety profile1

The safety of PEMAZYRE was evaluated in FIGHT-2031
The safety of PEMAZYRE was evaluated in 34 patients who were treated for MLNs with FGFR1 rearrangement. Patients were treated with PEMAZYRE 13.5 mg orally once daily on a continuous schedule (the approved recommended starting dosage) or for 14 days on followed by 7 days off therapy (an unapproved dosing regimen in MLNs with FGFR1 rearrangement) until disease progression, unacceptable toxicity, or they were able to receive allogeneic stem cell transplant. The median duration of treatment was 205 days (range: 30-1,347 days).1
- The most common (≥20%) adverse reactions were hyperphosphatemia, nail toxicity, alopecia, stomatitis, diarrhea, dry eye, fatigue, rash, abdominal pain, anemia, constipation, dry mouth, epistaxis, serous retinal detachment, extremity pain, decreased appetite, dry skin, dyspepsia, back pain, nausea, blurred vision, peripheral edema, and dizziness
- Serious adverse reactions occurred in 53% of patients receiving PEMAZYRE at all dosages. Serious adverse reactions in >5% of patients included acute kidney injury. Fatal adverse reactions occurred in 9% of patients who received PEMAZYRE, including acute kidney injury, multiple organ dysfunction syndrome, and malignant neoplasm progression, occurring in one patient each
- Permanent discontinuation due to an adverse reaction occurred in 12% of patients who received PEMAZYRE at all dosages. Adverse reactions requiring permanent discontinuation included cardiac failure, multiple organ dysfunction syndrome, blood alkaline phosphatase increase, and calciphylaxis
- In patients who started treatment on the recommended dosage (n=20), dosage interruptions and reductions occurred in 80% of patients
Adverse reactions (≥15%) in patients receiving PEMAZYRE1
PEMAZYRE N=34 Adverse Reaction | All Grades, %a | Grades 3 or 4, % |
|---|---|---|
| Metabolism and nutrition disorders | ||
| Hyperphosphatemiab | 74 | 2.9 |
| Decreased appetite | 24 | 6 |
| Skin and subcutaneous tissue disorders | ||
| Nail toxicityc | 62 | 21 |
| Alopecia | 59 | 0 |
| Rashd | 35 | 6 |
| Dry skine | 24 | 0 |
| Palmar-plantar erythrodysesthesiaf | 18 | 9 |
| Gastrointestinal disorders | ||
| Stomatitisg | 53 | 15 |
| Diarrhea | 50 | 2.9 |
| Abdominal painh | 35 | 2.9 |
| Constipation | 32 | 2.9 |
| Dry mouth | 32 | 0 |
| Dyspepsia | 24 | 0 |
| Nausea | 21 | 0 |
| Eye disorders | ||
| Dry eyei | 50 | 6 |
| Retinal pigment epithelial detachmentj | 26 | 0 |
| Vision blurred | 21 | 2.9 |
| Trichiasis | 18 | 2.9 |
| General disorders | ||
| Fatiguek | 44 | 9 |
| Edema peripheral | 21 | 0 |
| Pyrexia | 18 | 2.9 |
| Blood and lymphatic disorders | ||
| Anemia | 35 | 18 |
| Respiratory, thoracic, and mediastinal disorders | ||
| Epistaxis | 29 | 0 |
| Musculoskeletal and connective tissue disorders | ||
| Pain in extremity | 26 | 12 |
| Back painl | 24 | 9 |
| Nervous system disorders | ||
| Dizziness | 21 | 0 |
aGraded per NCI CTCAE v4.03.
bIncludes hyperphosphatemia and blood phosphorous increased; graded based on clinical severity and medical interventions taken according to the “investigations-other, specify” category in NCI CTCAE v4.03.
cIncludes ingrowing nail, nail bed inflammation, nail bed tenderness, nail discoloration, nail disorder, nail dystrophy, nail growth abnormal, nail infection, nail pigmentation, onychalgia, onychoclasis, onycholysis, onychomadesis, onychomycosis, and paronychia.
dIncludes dermatitis, dermatitis acneiform, lichen planus, rash, rash macular, and skin exfoliation.
eIncludes dry skin and xerosis.
fIncludes palmar erythema, palmar-plantar erythrodysesthesia, and plantar erythema.
gIncludes aphthous ulcer, cheilitis, lip ulceration, mouth ulceration, pharyngeal inflammation, stomatitis, and tongue ulceration.
hIncludes abdominal pain, abdominal pain lower, abdominal pain upper, and abdominal rigidity.
iIncludes dry eye, keratitis, lacrimation increased, meibomian gland dysfunction, and punctate keratitis.
jIncludes detachment of retinal pigment epithelium, maculopathy, retinal detachment, retinal disorder, retinal thickening, serous retinal detachment, and subretinal fluid.
kIncludes asthenia and fatigue.
lIncludes back pain and spinal pain.
Select laboratory abnormalities (≥20%) worsening from baseline in patients receiving PEMAZYRE1
PEMAZYRE N=34m Laboratory Abnormality | All Grades, %n | Grades 3 or 4, % |
|---|---|---|
| Hematology | ||
| Decreased lymphocytes | 65 | 16 |
| Decreased leukocytes | 65 | 15 |
| Decreased hemoglobin | 53 | 9 |
| Decreased neutrophils | 45 | 12 |
| Decreased platelets | 29 | 15 |
| Chemistry | ||
| Increased phosphateo | 97 | 2.9 |
| Increased alkaline phosphatase | 62 | 9 |
| Increased alanine aminotransferase | 50 | 12 |
| Increased aspartate aminotransferase | 47 | 9 |
| Increased creatininep | 44 | 0 |
| Decreased phosphate | 41 | 26 |
| Decreased sodium | 41 | 9 |
| Increased glucose | 33 | 3 |
| Decreased calcium | 26 | 2.9 |
| Increased calcium | 26 | 2.9 |
| Decreased potassium | 24 | 2.9 |
| Increased bilirubin | 21 | 0 |
mThe denominator used to calculate the rate varied from 31 to 34 based on the number of patients with a baseline value and at least one post-treatment value.
nGraded per NCI CTCAE v4.03.
oGraded per NCI CTCAE v5.0.
pBased on comparison to upper limit of normal.
Other clinically significant laboratory abnormalities: Prothrombin time/international normalized ratio was elevated in 16% (Grade 1 or 2 elevation) of patients. Uric acid was elevated in 18% of patients, including 2.9% with a Grade 3 or 4 elevation.1
RPED was observed in patients treated with PEMAZYRE1
Advise patients to inform you of any vision changes while taking PEMAZYRE
PEMAZYRE can cause retinal pigment epithelial detachment (RPED), which may cause symptoms such as blurred vision, visual floaters, or photopsia. Clinical trials of PEMAZYRE did not conduct routine monitoring, including optical coherence tomography (OCT), to detect asymptomatic RPED; therefore, the incidence of asymptomatic RPED with PEMAZYRE is unknown.1
- Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, RPED occurred in 11% of patients, including Grade 3-4 RPED in 1.3%1
- The median time to first onset of RPED was 56 days
- RPED led to dose interruption of PEMAZYRE in 3.1% of patients
- 1.3% of patients required dose reduction for RPED
- 0.2% of patients discontinued treatment due to RPED
- RPED resolved or improved to Grade 1 levels in 76% of patients who required dosage modification for RPED
Perform a comprehensive ophthalmological examination, including OCT, prior to initiation of PEMAZYRE, every 2 months for the first 6 months of treatment, and every 3 months thereafter during treatment. For onset of visual symptoms, refer patients for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of PEMAZYRE.
Modify the dose or permanently discontinue PEMAZYRE as recommended.
Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, dry eye occurred in 31% of patients, including Grade 3-4 in 1.6% of patients. Treat patients with ocular demulcents as needed.1
When to perform a comprehensive ophthalmological examination, including OCT1

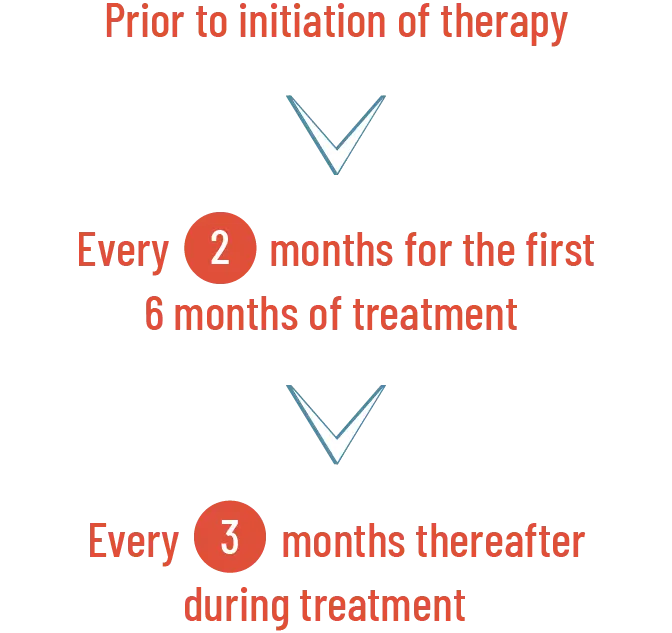
For onset of visual symptoms, refer patients for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of PEMAZYRE1
Dosage modifications for RPED1
- If asymptomatic and stable on serial examination, continue PEMAZYRE
- If symptomatic or worsening on serial examination, withhold PEMAZYRE
- If asymptomatic and improved on subsequent examination, resume PEMAZYRE at a lower dose
- If symptoms persist or no improvement is observed upon examination, consider permanent discontinuation of PEMAZYRE based on clinical status
Hyperphosphatemia was observed in patients treated with PEMAZYRE1
- PEMAZYRE can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcification, calcinosis, and non-uremic calciphylaxis. Increases in phosphate levels are a pharmacodynamic effect of PEMAZYRE
- Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, hyperphosphatemia was reported in 93% of patients based on laboratory values above the upper limit of normal
- The median time to onset of hyperphosphatemia was 8 days (range, 1-169)
- Phosphate-lowering therapy was required in 33% of patients receiving PEMAZYRE
Recommendations for management of hyperphosphatemia1
Monitor for hyperphosphatemia.
- Initiate a low-phosphate diet when serum phosphate level is >5.5 mg/dL
- For serum phosphate levels >7 mg/dL, initiate phosphate-lowering therapy and withhold, reduce the dose, or permanently discontinue PEMAZYRE based on duration and severity of hyperphosphatemia
Dose modifications for hyperphosphatemia1
| Severity | PEMAZYRE Dosage Modification |
|---|---|
| Serum phosphate >7 mg/dL to ≤10 mg/dL | Initiate phosphate-lowering therapy and monitor serum phosphate weekly. |
| Withhold PEMAZYRE if levels are not <7 mg/dL within 2 weeks of starting phosphate-lowering therapy. | |
| Resume PEMAZYRE at the same dose when phosphate levels are <7 mg/dL for first occurrence; resume at a lower dose level for subsequent recurrences. | |
| Serum phosphate >10 mg/dL | Initiate phosphate-lowering therapy and monitor serum phosphate weekly. |
| Withhold PEMAZYRE if levels are not ≤10 mg/dL within 1 week after starting phosphate-lowering therapy. | |
| Resume PEMAZYRE at the next lower dose level when phosphate levels are <7 mg/dL. | |
| Permanently discontinue PEMAZYRE for recurrence of serum phosphate >10 mg/dL following 2 dose reductions. |
Potential for embryo-fetal toxicity1
- Based on findings in an animal study and its mechanism of action, PEMAZYRE can cause fetal harm when administered to a pregnant woman
- Oral administration of pemigatinib to pregnant rats during the period of organogenesis caused fetal malformations, fetal growth retardation, and embryo-fetal death at maternal exposures lower than the human exposure based on area under the curve (AUC) at the clinical dose of 13.5 mg
Advise patients of potential risks1
Pregnant women
- Advise pregnant women of the potential risk to the fetus
Female patients
- Advise female patients of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose
- Advise female patients to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of pregnancy
- Advise patients not to breastfeed during treatment with PEMAZYRE and for 1 week after the last dose
Male patients
- Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose
CTCAE, Common Terminology Criteria for Adverse Events; FGFR, fibroblast growth factor receptor; MLN, myeloid/lymphoid neoplasm; NCI, National Cancer Institute.
Reference
- PEMAZYRE Prescribing Information. Wilmington, DE: Incyte Corporation.
Indications and Usage
PEMAZYRE® is indicated for the treatment of adults with relapsed or refractory myeloid/lymphoid neoplasms (MLNs) with fibroblast growth factor receptor 1 (FGFR1) rearrangement.
Important Safety Information
Ocular Toxicity
Retinal Pigment Epithelial Detachment (RPED): PEMAZYRE can cause RPED, which may cause symptoms such as blurred vision, visual floaters, or photopsia. Clinical trials of PEMAZYRE did not conduct routine monitoring including optical coherence tomography (OCT) to detect asymptomatic RPED; therefore, the incidence of asymptomatic RPED with PEMAZYRE is unknown.
Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, RPED occurred in 11% of patients, including Grade 3-4 RPED in 1.3%. The median time to first onset of RPED was 56 days. RPED led to dose interruption of PEMAZYRE in 3.1% of patients, and dose reduction and permanent discontinuation in 1.3% and in 0.2% of patients, respectively. RPED resolved or improved to Grade 1 levels in 76% of patients who required dosage modification of PEMAZYRE for RPED.
Perform a comprehensive ophthalmological examination including OCT prior to initiation of PEMAZYRE and every 2 months for the first 6 months and every 3 months thereafter during treatment. For onset of visual symptoms, refer patients for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of PEMAZYRE. Modify the dose or permanently discontinue PEMAZYRE as recommended in the prescribing information for PEMAZYRE.
Dry Eye: Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, dry eye occurred in 31% of patients, including Grade 3-4 in 1.6% of patients. Treat patients with ocular demulcents as needed.
Hyperphosphatemia and Soft Tissue Mineralization
PEMAZYRE can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcification, calcinosis, and non-uremic calciphylaxis. Increases in phosphate levels are a pharmacodynamic effect of PEMAZYRE. Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, hyperphosphatemia was reported in 93% of patients based on laboratory values above the upper limit of normal. The median time to onset of hyperphosphatemia was 8 days (range 1-169). Phosphate lowering therapy was required in 33% of patients receiving PEMAZYRE.
Monitor for hyperphosphatemia and initiate a low phosphate diet when serum phosphate level is >5.5 mg/dL. For serum phosphate levels >7 mg/dL, initiate phosphate lowering therapy and withhold, reduce the dose, or permanently discontinue PEMAZYRE based on duration and severity of hyperphosphatemia as recommended in the prescribing information.
Embryo-Fetal Toxicity
Based on findings in an animal study and its mechanism of action, PEMAZYRE can cause fetal harm when administered to a pregnant woman. Oral administration of pemigatinib to pregnant rats during the period of organogenesis caused fetal malformations, fetal growth retardation, and embryo-fetal death at maternal exposures lower than the human exposure based on area under the curve (AUC) at the clinical dose of 13.5 mg.
Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose.
Adverse Reactions: Myeloid/Lymphoid Neoplasms with FGFR1 Rearrangement
Serious adverse reactions occurred in 53% of patients receiving PEMAZYRE at all dosages (n=34). Serious adverse reactions in > 5% of patients included acute kidney injury. Fatal adverse reactions occurred in 9% of patients who received PEMAZYRE, including acute kidney injury, multiple organ dysfunction syndrome, and malignant neoplasm progression, occurring in one patient each.
Permanent discontinuation due to an adverse reaction occurred in 12% of patients who received PEMAZYRE at all dosages. Adverse reactions requiring permanent discontinuation included cardiac failure, multiple organ dysfunction syndrome, blood alkaline phosphatase increase, and calciphylaxis. In patients who started treatment on the recommended dosage (n = 20), adverse reactions requiring dosage interruption of PEMAZYRE occurred in 80% of patients. Adverse reactions which required dosage interruption in > 2 patients treated at the recommended dosage included nail toxicities (20%) and hyperphosphatemia (15%).
Dose reductions of PEMAZYRE due to an adverse reaction occurred in 80% of patients who started treatment on the recommended dosage. Adverse reactions requiring dose reductions occurring in > 2 patients were nail toxicities (20%), hyperphosphatemia (20%), and alopecia (15%).
Clinically relevant adverse reactions occurring in ≤10% of patients included fractures (2.1%). In all patients treated with pemigatinib, 0.5% experienced pathologic fractures (which included patients with and without cholangiocarcinoma [N = 635]). Soft tissue mineralization, including cutaneous calcification, calcinosis, and non-uremic calciphylaxis associated with hyperphosphatemia were observed with PEMAZYRE treatment.
Within the first 21-day cycle of PEMAZYRE dosing, serum creatinine increased (mean increase of 0.2 mg/dL) and reached steady state by Day 8, and then decreased during the 7 days off therapy. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
The most common (≥ 20%) adverse reactions were hyperphosphatemia (74%), nail toxicity (62%), alopecia (59%), stomatitis (53%), diarrhea (50%), dry eye (50%), fatigue (44%), rash (35%), abdominal pain (35%), anemia (35%), constipation (32%), dry mouth (32%), epistaxis (29%), retinal pigment epithelial detachment (26%), extremity pain (26%), decreased appetite (24%), dry skin (24%), dyspepsia (24%), back pain (24%), nausea (21%), blurred vision (21%), peripheral edema (21%), and dizziness (21%).
Drug Interactions
Avoid concomitant use of strong and moderate CYP3A inhibitors with PEMAZYRE. Reduce the dose of PEMAZYRE if concomitant use with a strong or moderate CYP3A inhibitor cannot be avoided. Avoid concomitant use of strong and moderate CYP3A inducers with PEMAZYRE.
Special Populations
Advise lactating women not to breastfeed during treatment with PEMAZYRE and for 1 week after the last dose.
Reduce the recommended dose of PEMAZYRE for patients with severe renal impairment as described in the prescribing information.
Reduce the recommended dose of PEMAZYRE for patients with severe hepatic impairment as described in the prescribing information.
Please see Full Prescribing Information for PEMAZYRE.

Indications and Usage
PEMAZYRE® is indicated for the treatment of adults with relapsed or refractory myeloid/lymphoid neoplasms (MLNs) with fibroblast growth factor receptor 1 (FGFR1) rearrangement.
Important Safety Information
Ocular Toxicity
Retinal Pigment Epithelial Detachment (RPED): PEMAZYRE can cause RPED, which may cause symptoms such as blurred vision, visual floaters, or photopsia. Clinical trials of PEMAZYRE did not conduct routine monitoring including optical coherence tomography (OCT) to detect asymptomatic RPED; therefore, the incidence of asymptomatic RPED with PEMAZYRE is unknown.
Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, RPED occurred in 11% of patients, including Grade 3-4 RPED in 1.3%. The median time to first onset of RPED was 56 days. RPED led to dose interruption of PEMAZYRE in 3.1% of patients, and dose reduction and permanent discontinuation in 1.3% and in 0.2% of patients, respectively. RPED resolved or improved to Grade 1 levels in 76% of patients who required dosage modification of PEMAZYRE for RPED.
Perform a comprehensive ophthalmological examination including OCT prior to initiation of PEMAZYRE and every 2 months for the first 6 months and every 3 months thereafter during treatment. For onset of visual symptoms, refer patients for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of PEMAZYRE. Modify the dose or permanently discontinue PEMAZYRE as recommended in the prescribing information for PEMAZYRE.
Dry Eye: Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, dry eye occurred in 31% of patients, including Grade 3-4 in 1.6% of patients. Treat patients with ocular demulcents as needed.
Hyperphosphatemia and Soft Tissue Mineralization
PEMAZYRE can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcification, calcinosis, and non-uremic calciphylaxis. Increases in phosphate levels are a pharmacodynamic effect of PEMAZYRE. Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, hyperphosphatemia was reported in 93% of patients based on laboratory values above the upper limit of normal. The median time to onset of hyperphosphatemia was 8 days (range 1-169). Phosphate lowering therapy was required in 33% of patients receiving PEMAZYRE.
Monitor for hyperphosphatemia and initiate a low phosphate diet when serum phosphate level is >5.5 mg/dL. For serum phosphate levels >7 mg/dL, initiate phosphate lowering therapy and withhold, reduce the dose, or permanently discontinue PEMAZYRE based on duration and severity of hyperphosphatemia as recommended in the prescribing information.
Embryo-Fetal Toxicity
Based on findings in an animal study and its mechanism of action, PEMAZYRE can cause fetal harm when administered to a pregnant woman. Oral administration of pemigatinib to pregnant rats during the period of organogenesis caused fetal malformations, fetal growth retardation, and embryo-fetal death at maternal exposures lower than the human exposure based on area under the curve (AUC) at the clinical dose of 13.5 mg.
Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose.
Adverse Reactions: Myeloid/Lymphoid Neoplasms with FGFR1 Rearrangement
Serious adverse reactions occurred in 53% of patients receiving PEMAZYRE at all dosages (n=34). Serious adverse reactions in > 5% of patients included acute kidney injury. Fatal adverse reactions occurred in 9% of patients who received PEMAZYRE, including acute kidney injury, multiple organ dysfunction syndrome, and malignant neoplasm progression, occurring in one patient each.
Permanent discontinuation due to an adverse reaction occurred in 12% of patients who received PEMAZYRE at all dosages. Adverse reactions requiring permanent discontinuation included cardiac failure, multiple organ dysfunction syndrome, blood alkaline phosphatase increase, and calciphylaxis. In patients who started treatment on the recommended dosage (n = 20), adverse reactions requiring dosage interruption of PEMAZYRE occurred in 80% of patients. Adverse reactions which required dosage interruption in > 2 patients treated at the recommended dosage included nail toxicities (20%) and hyperphosphatemia (15%).
Dose reductions of PEMAZYRE due to an adverse reaction occurred in 80% of patients who started treatment on the recommended dosage. Adverse reactions requiring dose reductions occurring in > 2 patients were nail toxicities (20%), hyperphosphatemia (20%), and alopecia (15%).
Clinically relevant adverse reactions occurring in ≤10% of patients included fractures (2.1%). In all patients treated with pemigatinib, 0.5% experienced pathologic fractures (which included patients with and without cholangiocarcinoma [N = 635]). Soft tissue mineralization, including cutaneous calcification, calcinosis, and non-uremic calciphylaxis associated with hyperphosphatemia were observed with PEMAZYRE treatment.
Within the first 21-day cycle of PEMAZYRE dosing, serum creatinine increased (mean increase of 0.2 mg/dL) and reached steady state by Day 8, and then decreased during the 7 days off therapy. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
The most common (≥ 20%) adverse reactions were hyperphosphatemia (74%), nail toxicity (62%), alopecia (59%), stomatitis (53%), diarrhea (50%), dry eye (50%), fatigue (44%), rash (35%), abdominal pain (35%), anemia (35%), constipation (32%), dry mouth (32%), epistaxis (29%), retinal pigment epithelial detachment (26%), extremity pain (26%), decreased appetite (24%), dry skin (24%), dyspepsia (24%), back pain (24%), nausea (21%), blurred vision (21%), peripheral edema (21%), and dizziness (21%).
Drug Interactions
Avoid concomitant use of strong and moderate CYP3A inhibitors with PEMAZYRE. Reduce the dose of PEMAZYRE if concomitant use with a strong or moderate CYP3A inhibitor cannot be avoided. Avoid concomitant use of strong and moderate CYP3A inducers with PEMAZYRE.
Special Populations
Advise lactating women not to breastfeed during treatment with PEMAZYRE and for 1 week after the last dose.
Reduce the recommended dose of PEMAZYRE for patients with severe renal impairment as described in the prescribing information.
Reduce the recommended dose of PEMAZYRE for patients with severe hepatic impairment as described in the prescribing information.
Please see Full Prescribing Information for PEMAZYRE.