For US Healthcare Professionals Only
Managing Adverse Reactions
SELECT ADVERSE REACTIONS RELATED TO FGFR INHIBITION1
PEMAZYRE targets and inhibits FGFR1, 2, and 32



Retinal pigment epithelial detachment (RPED)
Monitoring
Advise patients to inform you of any vision changes while taking PEMAZYRE.2
Perform a comprehensive ophthalmological examination, including optical coherence tomography (OCT), prior to initiation of PEMAZYRE, every 2 months for the first 6 months of treatment, and every 3 months thereafter during treatment.2
OCT is a non-invasive imaging test that uses light waves to map and measure the thickness of distinctive layers of the retina.4
When to perform a comprehensive ophthalmological examination, including OCT2


Management
- For onset of visual symptoms, refer patient for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of PEMAZYRE2
- Modify the dose or permanently discontinue PEMAZYRE as recommended2
Recommended dose modification of PEMAZYRE for RPED2

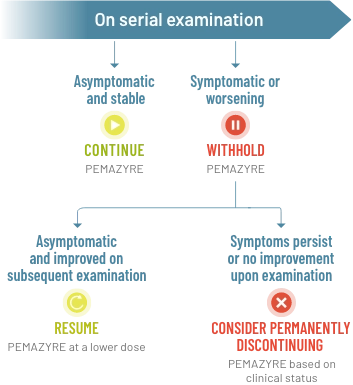

Management of RPED in clinical trials
Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, RPED occurred in 11% of patients, including Grade 3-4 RPED in 1.3%.2
The median time to first onset of RPED was 56 days
Dose modifications:
- RPED resolved or improved to Grade 1 levels in 76% of patients who required dosage modification for RPED
- 1.3% of patients required dose reduction for RPED
- RPED led to dose interruption of PEMAZYRE in 3.1% of patients
- 0.2% of patients permanently discontinued treatment due to RPED
Clinical trials of PEMAZYRE did not conduct routine monitoring, including OCT, to detect asymptomatic RPED; therefore, the incidence of asymptomatic RPED with PEMAZYRE is unknown.2
Scientific Background
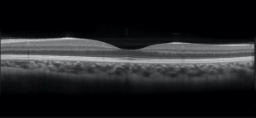
OCT image of normal retina. Reprinted with permission.5
FGF/FGFR and the retinal epithelium5
The FGF/FGFR pathway plays an essential role in the retinal epithelium.
Fibroblast growth factor (FGF) is a neurotrophic factor found in the retinal pigment epithelium (RPE)
FGF promotes growth in immature RPE cells and helps prevent apoptosis of mature cells
FGFR-mitogen-activated protein kinase (MAPK) signaling helps protect the RPE from injury
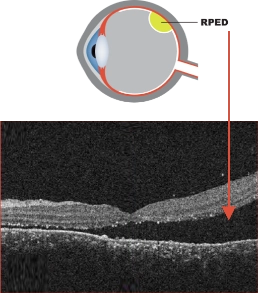
OCT image of exudative retinal detachment.7
Adapted from Simunovic MP, et al. BMC Ophthalmol. 2020;20(1):349.
OCT image of exudative retinal detachment.7 Adapted from Simunovic MP, et al. BMC Ophthalmol. 2020;20(1):349.
RPED may be a related effect of FGFR inhibition1
RPED is characterized by the accumulation of fluid under the retina.6
RPED does not represent an actual, physical tear or separation of the retina5
Treatment of RPED is conservative; it often resolves within days without requiring treatment5
PEMAZYRE is an FGFR 1, 2, and 3 inhibitor and therefore can cause RPED; this may cause symptoms such as blurred vision, visual floaters, or photopsia.1,2,5
Hyperphosphatemia and soft tissue mineralization
Monitoring
Monitor for hyperphosphatemia. Inform patients that they may experience an increase in phosphate levels and of the need to monitor serum phosphate levels. Advise patients to immediately inform their healthcare provider of any symptoms related to acute change in phosphate levels, such as muscle cramps, numbness, or tingling around the mouth.2
Management
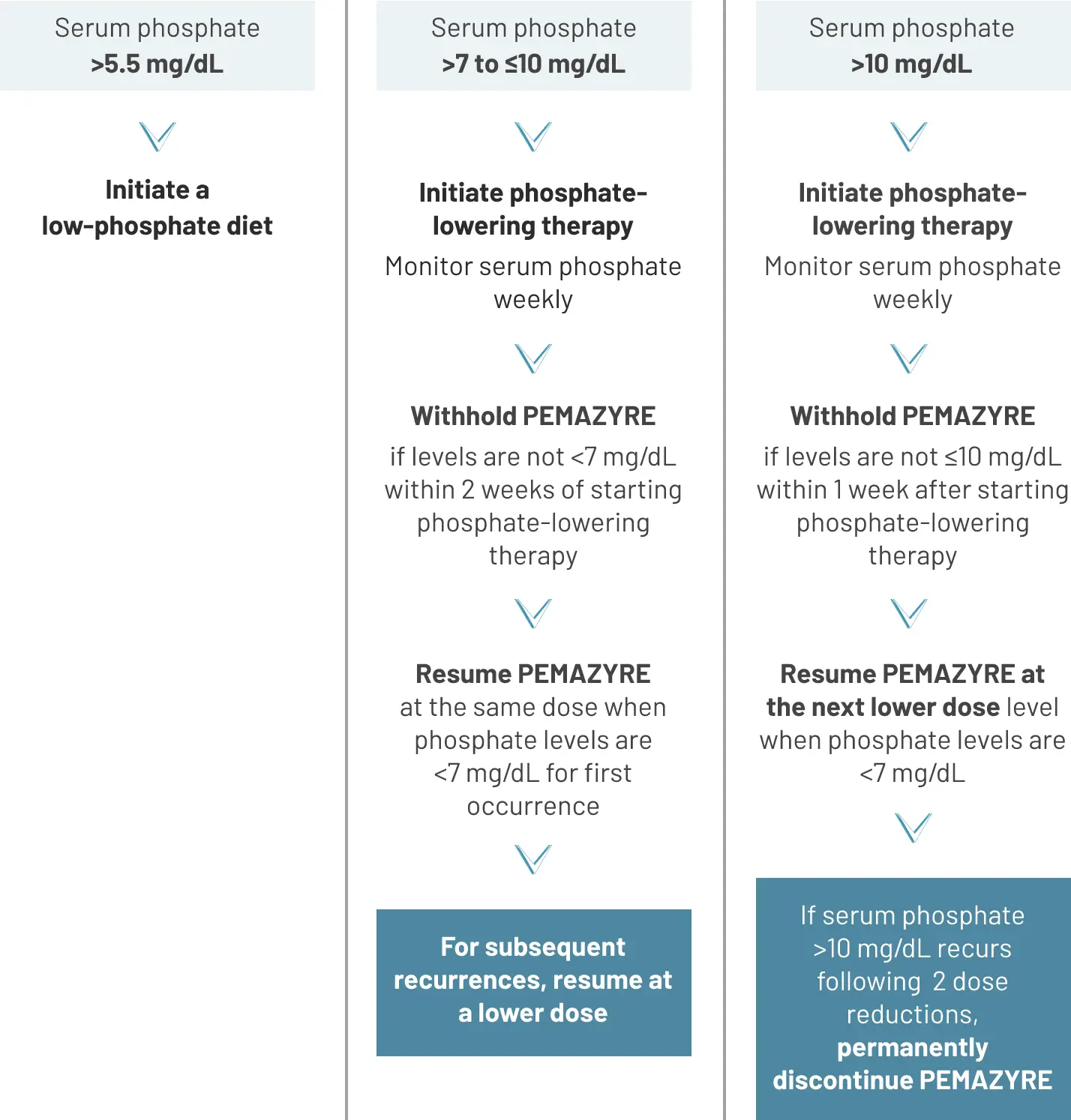
Recommended dose modification2
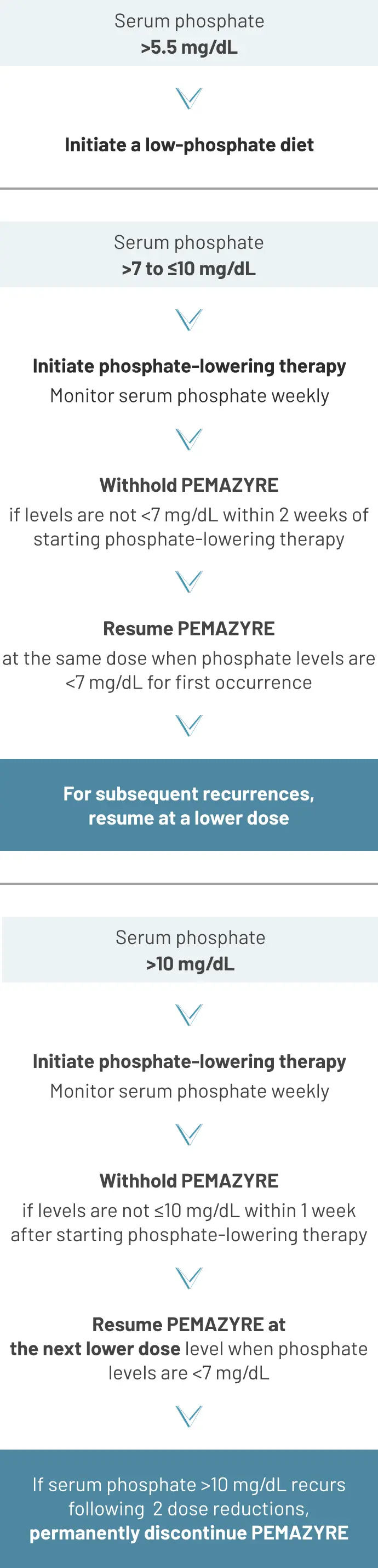
Management of hyperphosphatemia in clinical trials
Among patients taking PEMAZYRE in the FIGHT-202 study (N=146)2:
All cases of hyperphosphatemia were Grade 1 or 22
Phosphate binders were used by 27 of 146 patients (18%) and diuretics by 1 of 146 patients (1%)8
In the FIGHT-202 study, serum phosphate was measured on Day 1 of every cycle following a 1-week dosing holiday, starting with the second cycle of treatment with PEMAZYRE.9
Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials2:
Hyperphosphatemia was reported in 93% of patients based on laboratory values above the upper limit of normal
The median time to onset of hyperphosphatemia was 8 days (range, 1-169)
Phosphate-lowering therapy was required in 33% of patients receiving PEMAZYRE
Scientific Background
Increase in phosphate levels is a related effect of FGFR inhibition8,10,11
FGF23, an FGFR ligand, is involved in phosphate homeostasis by reducing uptake of phosphate by the kidney.
FGFR inhibition may lead to blockade of FGF23 signaling and in turn cause hyperphosphatemia.
Hyperphosphatemia is often asymptomatic12
Hyperphosphatemia can be associated with soft tissue and organ calcification3
Dermatologic/mucosal adverse reactions13
Nail toxicity
Stomatitis
Alopecia
Management
In the FIGHT-202 study, examples of nail toxicity included2:
Onychomadesis
Paronychia
Onycholysis
Nail hypertrophy
Nail dystrophy
Nail discoloration
Consider counseling and education on the potential for nail changes before initiation of treatment.13
Preventative strategies may include avoidance of13:
- Prolonged contact with water
- Repeated trauma
- Friction
- Pressure on the nails and nail beds
Patients may be advised to13:
- Limit the use of nail polish removers or hardeners
- Avoid biting nails or cutting nails too short
- Use topical emollients
- Wear loose-fitting socks and footwear
Preventative correction of nail curvature may be considered.13
Management of nail toxicity in clinical trials
Among patients taking PEMAZYRE in the FIGHT-202 study (N=146)2:
43% experienced nail toxicity2
- 2.1% experienced Grade 3 or 4 nail toxicity
Dose modifications8:
- 3% required dose reduction for nail toxicity
- 4% required dose interruption for nail toxicity
Consider counseling and education on the potential for stomatitis before initiation of treatment.13
Preventative strategies may include13:
- Dental work to address tooth and gum disease before start of treatment
- Education regarding the importance of thorough and frequent cleaning of the oral cavity
Patients may be advised to13:
- Avoid salty, spicy, or citrus-based foods
- Avoid hot beverages
Potential management approaches may include use of14:
- Coating agents, such as bismuth salicylate, sucralfate, or other antacids
- Water-soluble mouth/lip lubricants
- “Magic mouthwash” (may include antifungals, antibacterials, steroids, and/or local anesthetics)
- Topical or oral analgesics
- Topical anesthetics
Management of stomatitis in clinical trials
Among patients taking PEMAZYRE in the FIGHT-202 study (N=146)2:
35% experienced stomatitis2
- 5% experienced Grade 3 or 4 stomatitis
Dose modifications8:
- 5 patients required dose reductions for stomatitis
- 11 patients required dose interruptions for stomatitis
Preventative measures normally considered for patients undergoing traditional chemotherapy (eg, scalp compression, scalp cooling, medications) may not be applicable.13
Management approaches to consider may include13:
- Prophylactic or reactive topical medications for the scalp to encourage hair regrowth
- High-potency topical corticosteroids
- Camouflaging methods that create the appearance of naturally fuller hair
Attention should be focused on early identification and management.13
Management of alopecia in clinical trials
Among patients taking PEMAZYRE in the FIGHT-202 study (N=146)2:
49% experienced alopecia
- No patients experienced Grade 3 or 4 alopecia
Scientific Background
FGFR inhibition and dermatologic events13
Nail toxicity, stomatitis, and alopecia have been observed with FGFR inhibition.
The pathophysiological mechanisms of these events are not fully understood.
Several possible mechanisms have been proposed, including:
- Inhibition of FGFR in keratinocytes, inducing dysregulation of hair-follicle homeostasis and epidermal proliferation
- Inhibition of hormonal FGF signaling by FGF19, FGF21, and FGF23
For additional information about adverse reactions and dose modifications, please see the Full Prescribing Information for PEMAZYRE.
Important Safety Information
Ocular Toxicity
Retinal Pigment Epithelial Detachment (RPED): PEMAZYRE can cause RPED, which may cause symptoms such as blurred vision, visual floaters, or photopsia. Clinical trials of PEMAZYRE did not conduct routine monitoring including optical coherence tomography (OCT) to detect asymptomatic RPED; therefore, the incidence of asymptomatic RPED with PEMAZYRE is unknown.
Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, RPED occurred in 11% of patients, including Grade 3-4 RPED in 1.3%. The median time to first onset of RPED was 56 days. RPED led to dose interruption of PEMAZYRE in 3.1% of patients, and dose reduction and permanent discontinuation in 1.3% and in 0.2% of patients, respectively. RPED resolved or improved to Grade 1 levels in 76% of patients who required dosage modification of PEMAZYRE for RPED.
Perform a comprehensive ophthalmological examination including OCT prior to initiation of PEMAZYRE and every 2 months for the first 6 months and every 3 months thereafter during treatment. For onset of visual symptoms, refer patients for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of PEMAZYRE. Modify the dose or permanently discontinue PEMAZYRE as recommended in the prescribing information for PEMAZYRE.
Dry Eye: Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, dry eye occurred in 31% of patients, including Grade 3-4 in 1.6% of patients. Treat patients with ocular demulcents as needed.
Hyperphosphatemia and Soft Tissue Mineralization
PEMAZYRE can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcification, calcinosis, and non-uremic calciphylaxis. Increases in phosphate levels are a pharmacodynamic effect of PEMAZYRE. Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, hyperphosphatemia was reported in 93% of patients based on laboratory values above the upper limit of normal. The median time to onset of hyperphosphatemia was 8 days (range 1-169). Phosphate lowering therapy was required in 33% of patients receiving PEMAZYRE.
Monitor for hyperphosphatemia and initiate a low phosphate diet when serum phosphate level is >5.5 mg/dL. For serum phosphate levels >7 mg/dL, initiate phosphate lowering therapy and withhold, reduce the dose, or permanently discontinue PEMAZYRE based on duration and severity of hyperphosphatemia as recommended in the prescribing information.
Embryo-Fetal Toxicity
Based on findings in an animal study and its mechanism of action, PEMAZYRE can cause fetal harm when administered to a pregnant woman. Oral administration of pemigatinib to pregnant rats during the period of organogenesis caused fetal malformations, fetal growth retardation, and embryo-fetal death at maternal exposures lower than the human exposure based on area under the curve (AUC) at the clinical dose of 13.5 mg.
Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose.
Adverse Reactions: Cholangiocarcinoma
Serious adverse reactions occurred in 45% of patients receiving PEMAZYRE (n=146). Serious adverse reactions in ≥2% of patients who received PEMAZYRE included abdominal pain, pyrexia, cholangitis, pleural effusion, acute kidney injury, cholangitis infective, failure to thrive, hypercalcemia, hyponatremia, small intestinal obstruction, and urinary tract infection. Fatal adverse reactions occurred in 4.1% of patients, including failure to thrive, bile duct obstruction, cholangitis, sepsis, and pleural effusion.
Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received PEMAZYRE. Adverse reactions requiring permanent discontinuation in ≥1% of patients included intestinal obstruction and acute kidney injury.
Dosage interruptions due to an adverse reaction occurred in 43% of patients who received PEMAZYRE. Adverse reactions requiring dosage interruption in ≥1% of patients included stomatitis, palmar-plantar erythrodysesthesia syndrome, arthralgia, fatigue, abdominal pain, AST increased, asthenia, pyrexia, ALT increased, cholangitis, small intestinal obstruction, alkaline phosphatase increased, diarrhea, hyperbilirubinemia, electrocardiogram QT prolonged, decreased appetite, dehydration, hypercalcemia, hyperphosphatemia, hypophosphatemia, back pain, pain in extremity, syncope, acute kidney injury, onychomadesis, and hypotension.
Dose reductions due to an adverse reaction occurred in 14% of patients who received PEMAZYRE. Adverse reactions requiring dosage reductions in ≥1% of patients who received PEMAZYRE included stomatitis, arthralgia, palmar-plantar erythrodysesthesia syndrome, asthenia, and onychomadesis.
Clinically relevant adverse reactions occurring in ≤10% of patients included fractures (2.1%). In all patients treated with pemigatinib, 0.5% experienced pathologic fractures (which included patients with and without cholangiocarcinoma [N = 635]). Soft tissue mineralization, including cutaneous calcification, calcinosis, and non-uremic calciphylaxis associated with hyperphosphatemia were observed with PEMAZYRE treatment.
Within the first 21-day cycle of PEMAZYRE dosing, serum creatinine increased (mean increase of 0.2 mg/dL) and reached steady state by Day 8, and then decreased during the 7 days off therapy. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
In cholangiocarcinoma (n=146) the most common adverse reactions (incidence ≥20%) were hyperphosphatemia (60%), alopecia (49%), diarrhea (47%), nail toxicity (43%), fatigue (42%), dysgeusia (40%), nausea (40%), constipation (35%), stomatitis (35%), dry eye (35%), dry mouth (34%), decreased appetite (33%), vomiting (27%), arthralgia (25%), abdominal pain (23%), hypophosphatemia (23%), back pain (20%), and dry skin (20%).
Drug Interactions
Avoid concomitant use of strong and moderate CYP3A inhibitors with PEMAZYRE. Reduce the dose of PEMAZYRE if concomitant use with a strong or moderate CYP3A inhibitor cannot be avoided. Avoid concomitant use of strong and moderate CYP3A inducers with PEMAZYRE.
Special Populations
Advise lactating women not to breastfeed during treatment with PEMAZYRE and for 1 week after the last dose.
Reduce the recommended dose of PEMAZYRE for patients with severe renal impairment as described in the prescribing information.
Reduce the recommended dose of PEMAZYRE for patients with severe hepatic impairment as described in the prescribing information.
Please see Full Prescribing Information for PEMAZYRE.
Indications and Usage
PEMAZYRE® is indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).

Important Safety Information
Ocular Toxicity
Retinal Pigment Epithelial Detachment (RPED): PEMAZYRE can cause RPED, which may cause symptoms such as blurred vision, visual floaters, or photopsia. Clinical trials of PEMAZYRE did not conduct routine monitoring including optical coherence tomography (OCT) to detect asymptomatic RPED; therefore, the incidence of asymptomatic RPED with PEMAZYRE is unknown.
Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, RPED occurred in 11% of patients, including Grade 3-4 RPED in 1.3%. The median time to first onset of RPED was 56 days. RPED led to dose interruption of PEMAZYRE in 3.1% of patients, and dose reduction and permanent discontinuation in 1.3% and in 0.2% of patients, respectively. RPED resolved or improved to Grade 1 levels in 76% of patients who required dosage modification of PEMAZYRE for RPED.
Perform a comprehensive ophthalmological examination including OCT prior to initiation of PEMAZYRE and every 2 months for the first 6 months and every 3 months thereafter during treatment. For onset of visual symptoms, refer patients for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of PEMAZYRE. Modify the dose or permanently discontinue PEMAZYRE as recommended in the prescribing information for PEMAZYRE.
Dry Eye: Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, dry eye occurred in 31% of patients, including Grade 3-4 in 1.6% of patients. Treat patients with ocular demulcents as needed.
Hyperphosphatemia and Soft Tissue Mineralization
PEMAZYRE can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcification, calcinosis, and non-uremic calciphylaxis. Increases in phosphate levels are a pharmacodynamic effect of PEMAZYRE. Among 635 patients who received a starting dose of PEMAZYRE 13.5 mg across clinical trials, hyperphosphatemia was reported in 93% of patients based on laboratory values above the upper limit of normal. The median time to onset of hyperphosphatemia was 8 days (range 1-169). Phosphate lowering therapy was required in 33% of patients receiving PEMAZYRE.
Monitor for hyperphosphatemia and initiate a low phosphate diet when serum phosphate level is >5.5 mg/dL. For serum phosphate levels >7 mg/dL, initiate phosphate lowering therapy and withhold, reduce the dose, or permanently discontinue PEMAZYRE based on duration and severity of hyperphosphatemia as recommended in the prescribing information.
Embryo-Fetal Toxicity
Based on findings in an animal study and its mechanism of action, PEMAZYRE can cause fetal harm when administered to a pregnant woman. Oral administration of pemigatinib to pregnant rats during the period of organogenesis caused fetal malformations, fetal growth retardation, and embryo-fetal death at maternal exposures lower than the human exposure based on area under the curve (AUC) at the clinical dose of 13.5 mg.
Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMAZYRE and for 1 week after the last dose.
Adverse Reactions: Cholangiocarcinoma
Serious adverse reactions occurred in 45% of patients receiving PEMAZYRE (n=146). Serious adverse reactions in ≥2% of patients who received PEMAZYRE included abdominal pain, pyrexia, cholangitis, pleural effusion, acute kidney injury, cholangitis infective, failure to thrive, hypercalcemia, hyponatremia, small intestinal obstruction, and urinary tract infection. Fatal adverse reactions occurred in 4.1% of patients, including failure to thrive, bile duct obstruction, cholangitis, sepsis, and pleural effusion.
Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received PEMAZYRE. Adverse reactions requiring permanent discontinuation in ≥1% of patients included intestinal obstruction and acute kidney injury.
Dosage interruptions due to an adverse reaction occurred in 43% of patients who received PEMAZYRE. Adverse reactions requiring dosage interruption in ≥1% of patients included stomatitis, palmar-plantar erythrodysesthesia syndrome, arthralgia, fatigue, abdominal pain, AST increased, asthenia, pyrexia, ALT increased, cholangitis, small intestinal obstruction, alkaline phosphatase increased, diarrhea, hyperbilirubinemia, electrocardiogram QT prolonged, decreased appetite, dehydration, hypercalcemia, hyperphosphatemia, hypophosphatemia, back pain, pain in extremity, syncope, acute kidney injury, onychomadesis, and hypotension.
Dose reductions due to an adverse reaction occurred in 14% of patients who received PEMAZYRE. Adverse reactions requiring dosage reductions in ≥1% of patients who received PEMAZYRE included stomatitis, arthralgia, palmar-plantar erythrodysesthesia syndrome, asthenia, and onychomadesis.
Clinically relevant adverse reactions occurring in ≤10% of patients included fractures (2.1%). In all patients treated with pemigatinib, 0.5% experienced pathologic fractures (which included patients with and without cholangiocarcinoma [N = 635]). Soft tissue mineralization, including cutaneous calcification, calcinosis, and non-uremic calciphylaxis associated with hyperphosphatemia were observed with PEMAZYRE treatment.
Within the first 21-day cycle of PEMAZYRE dosing, serum creatinine increased (mean increase of 0.2 mg/dL) and reached steady state by Day 8, and then decreased during the 7 days off therapy. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
In cholangiocarcinoma (n=146) the most common adverse reactions (incidence ≥20%) were hyperphosphatemia (60%), alopecia (49%), diarrhea (47%), nail toxicity (43%), fatigue (42%), dysgeusia (40%), nausea (40%), constipation (35%), stomatitis (35%), dry eye (35%), dry mouth (34%), decreased appetite (33%), vomiting (27%), arthralgia (25%), abdominal pain (23%), hypophosphatemia (23%), back pain (20%), and dry skin (20%).
Drug Interactions
Avoid concomitant use of strong and moderate CYP3A inhibitors with PEMAZYRE. Reduce the dose of PEMAZYRE if concomitant use with a strong or moderate CYP3A inhibitor cannot be avoided. Avoid concomitant use of strong and moderate CYP3A inducers with PEMAZYRE.
Special Populations
Advise lactating women not to breastfeed during treatment with PEMAZYRE and for 1 week after the last dose.
Reduce the recommended dose of PEMAZYRE for patients with severe renal impairment as described in the prescribing information.
Reduce the recommended dose of PEMAZYRE for patients with severe hepatic impairment as described in the prescribing information.
Please see Full Prescribing Information for PEMAZYRE.
Indications and Usage
PEMAZYRE® is indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).